Learn about the rare disease called: Apert Syndrome
Apert syndrome, also known as localized early hard bone disease, and narrow airway is a rare but very malignant disease and causes serious illness in children.
This syndrome appears to be due to variability in semen . This transformation prevents the development of the mind, causing the toes and fingers to stick together and stimulating the division of the cell chain to form semen.
1 in 70,000 children has congenital Apert. The change in DNA appears on a single gene on the baby chromosome 10 and is closely related to the age of the father. This syndrome causes the baby's body to stunt and lead to a lack of cartilage development, whereby soft organs or organs are developing normally but the bones are not growing.
The cause of Apert Syndrome
Apert syndrome is caused by a rare mutation in a single gene. This mutant gene is normally responsible for matching bones at the right time during development.
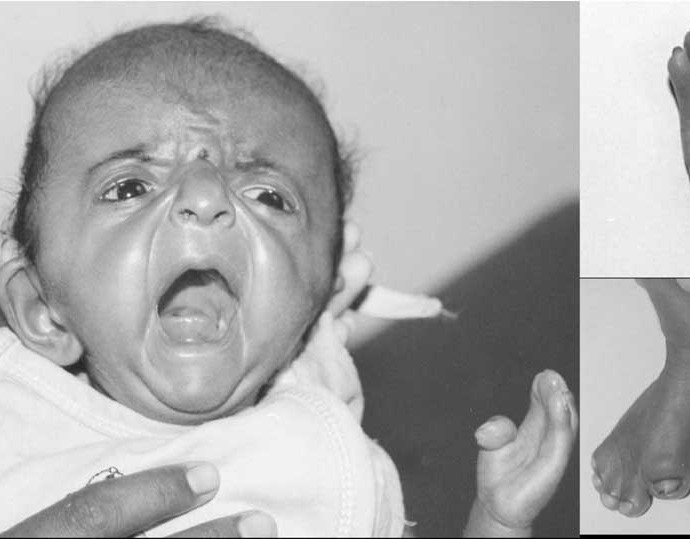
A baby has Apert syndrome or early local hard bone disease.
Apert syndrome is a common chromosomal disorder , about two-thirds of cases are caused by a C-mutation in G at position 755 in the FGFR2 gene, causing a change from Ser to TRP in protein.
In most cases, mutated Apert gene syndrome seems to be random. Only about one in 65,000 babies are born with Apert syndrome. Apert syndrome can be inherited from infected adults with a probability of 50%, or may be due to spontaneous mutations.
Symptoms of Apert Syndrome
The defective gene in newborns with Apert syndrome causes skull bones to be pinched by the closed skull early, a process called craniosynostosis . The brain continues to grow inside an unusually narrow skull, putting pressure on the skull and face bones. Abnormal skull and facial growth in Apert syndrome produce the following major signs and symptoms:
- Pyramid skull due to early skull joints.
- Sunken eyes, often with poor eyelids closed, muscle movements are unbalanced.
- A sunken face. Poor development of the middle part of the face leads to low and protruding cheekbones.
Other symptoms also result from abnormal skull growth:
- There may be mental retardation (in most children with Apert syndrome).
- Obstruction causes sleep apnea.
- Hearing loss due to ear infections, sinusitis, hearing loss.
- Stick bones of fingers and legs (syndactyly) - with hands or feet. Some children with Apert syndrome also have heart defects, digestion, or urinary system problems that are not seen in other craniofacial premature synaptic syndromes.
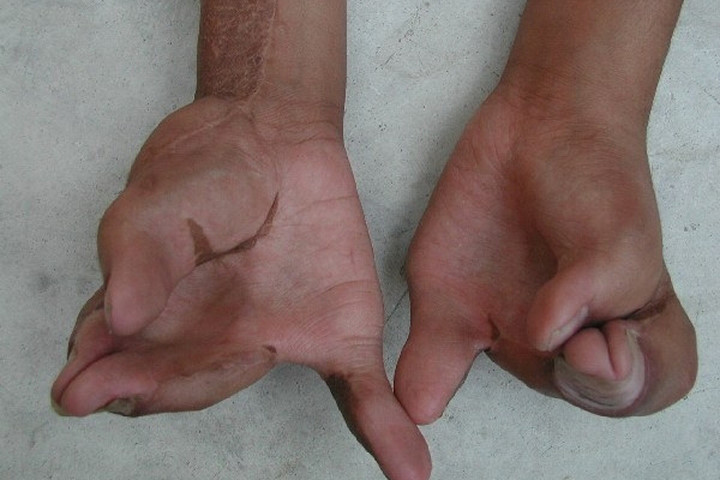
The fingers are stuck together and cramped.
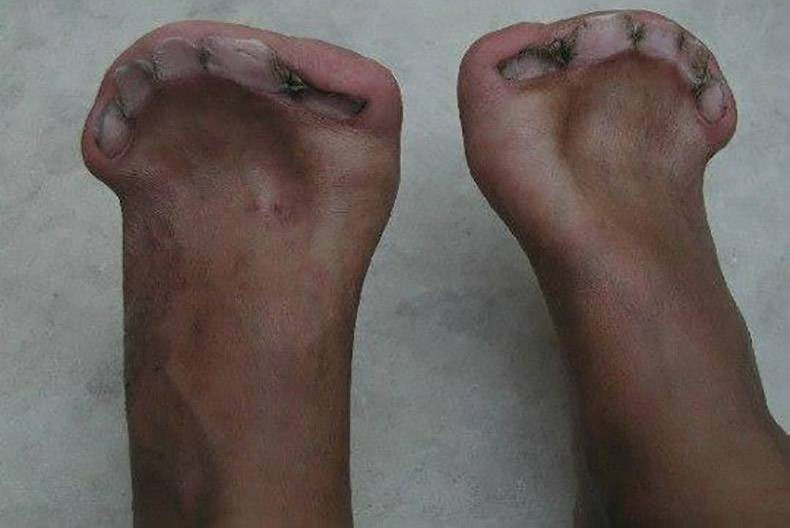
Malformed feet with 5 toenails but completely without fingers.
Diagnosis of Apert Syndrome
Doctors often suspect Apert syndrome or other neonatal craniosynostosis syndrome because of the infant's appearance. Genetic testing can often identify Apert syndrome or other causes of abnormal skull formation.
Treatment of Apert Syndrome
Children with Apert syndrome may have abnormal pronunciation. There are usually no nasal sounds because the part between the underdeveloped face, the small nose, and the soft palate is very long. If there is cleft palate, patients with Apert syndrome can also pronounce without nasal sounds. The pronunciation is often unclear due to the occlusive bite and the protruding palate. Poor hearing or overall delay may also affect language development and pronunciation. Surgery to repair abnormal connections between bones is the main treatment for Apert syndrome.
The best time to release the joints of the skull joints is about three to six months old ; may take up to 18 months of age. Early surgery allows the young brain to have space to grow. In addition to releasing the joints of the skull, deformed brow bone and other bones will be repositioned to correct protrusion and deformity of the upper part of the face. A plastic surgeon and a neurosurgeon will combine surgery to deliver the most effective operation. If the middle part of the face and the upper part of the jaw does not grow properly, the surgery should be continued when the child is older. Hand sticking is often separated in the early years of life to make hands more straight and function better. Sometimes, surgery may be required so that the child has full functioning.
Surgery for Apert syndrome takes place in three steps:
- Release craniosynostosis (craniosynostosis) . An abnormal bone splitting skull surgeon and partially rearranging some of them. This surgery is usually done when a child is between 6 and 8 months old.
- Surgery to improve the midface (Midface advancement) . When the child develops with Apert syndrome, the facial bone becomes skewed. The surgeon cuts the jaw and cheekbones and repositiones these bones normally. Surgery can be done at any time from 4 to 12 years of age. Orthopedic surgery may be needed additionally, especially when the midface advancement is performed early when the child is young.
- Repair wide eyes (hypertelorism) . The surgeon will remove a wedge of bone in the skull between the eyes, bring the eye sockets closer together, adjust the jaw in conjunction with this surgery.
- 14 most bizarre diseases in the world
- The 20 most unusual and rare diseases in the world
- Strange diseases
- Strange disease turns a young woman into a
- The disease syndrome makes people special
- The most feared strange diseases in the world
- Strange disease: The boy has a face
- Strange diseases discovered in the 21st century
- Williams syndrome, the disease is too friendly
- The syndrome of 'bad fear' can ruin a lifetime
- The girl with the syndrome of
- Learn Down syndrome and the truth you should know
 Green tea cleans teeth better than mouthwash?
Green tea cleans teeth better than mouthwash? Death kiss: This is why you should not let anyone kiss your baby's lips
Death kiss: This is why you should not let anyone kiss your baby's lips What is salmonellosis?
What is salmonellosis? Caution should be exercised when using aloe vera through eating and drinking
Caution should be exercised when using aloe vera through eating and drinking